Arkivi Terapeutik Nr. 03 2018 - Hemokromatoza - gjendja aktuale e problemit

Hemokromatoza është një patologji trashëgimore e shoqëruar me thithjen e lartë të hekurit në organet e tretjes dhe akumulimin e saj të tepërt pasues në organe të ndryshme të brendshme.
Mëlçia vuan më shumë se të tjerët. Zbulimi i hershëm i hemokromatozës, diagnoza dhe trajtimi i saj nuk do të lejojnë zhvillimin e pasojave.
Hemokromatoza - gjendje moderne e problemit
N.B. VOLOSHINА1, M.F. OSIPENKO1, N.V. LITVINOVA1, A.N.VOLOSHIN2
1 Universiteti Shtetëror Mjekësor Novosibirsk FGBOU në NSMU të Ministrisë së Shëndetësisë të Rusisë, Rusisë,
Spitali Klinik 2Novosibirsk City 2, Rusi
Sindroma e mbingarkesës së hekurit mund të shoqërohet me gjendje të ndryshme të fituara dhe faktorë trashëgues. Hemokromatoza e trashëguar është çrregullimi gjenetik më i zakonshëm. Pa ndërhyrjen terapeutike sëmundja mund të çojë në zhvillimin e komplikimeve të rrezikshme për jetën, si cirroza, karcinoma hepatocelulare. Artikulli paraqet të dhëna për patogjenezën, diagnozën dhe trajtimin e hemokromatozës trashëgimore. Jepet vëzhgimi klinik vetanak.
Keywords: hemokromatoza trashëgimore, trajtimi, flebotomia.
 Hemokromatoza është një sëmundje që shoqërohet me akumulimin e niveleve të larta patologjike të hekurit në trup, gjë që çon në çrregullime funksionale të disa organeve. Në mënyrë tipike, thithja e hekurit rregullohet fort, si rezultat i së cilës trupi nuk është në gjendje të sekretojë hekurin e tepërt. Hekuri i tepërt grumbullohet në qelizat si hemosiderina. Kjo përfundimisht çon në vdekjen e qelizave dhe zëvendësimin e këtyre qelizave me ind fibroze, gjë që çon në prishjen e strukturës dhe funksionimit të organeve. Me hemokromatozë është e mundur dëmtimi i mëlçisë, pankreasit, zemrës, gjëndrës tiroide, nyjeve, lëkurës, gonadave dhe gjëndrrës së hipofizës.
Hemokromatoza është një sëmundje që shoqërohet me akumulimin e niveleve të larta patologjike të hekurit në trup, gjë që çon në çrregullime funksionale të disa organeve. Në mënyrë tipike, thithja e hekurit rregullohet fort, si rezultat i së cilës trupi nuk është në gjendje të sekretojë hekurin e tepërt. Hekuri i tepërt grumbullohet në qelizat si hemosiderina. Kjo përfundimisht çon në vdekjen e qelizave dhe zëvendësimin e këtyre qelizave me ind fibroze, gjë që çon në prishjen e strukturës dhe funksionimit të organeve. Me hemokromatozë është e mundur dëmtimi i mëlçisë, pankreasit, zemrës, gjëndrës tiroide, nyjeve, lëkurës, gonadave dhe gjëndrrës së hipofizës.
Mbingarkesa e hekurit, e cila shkakton hemokromatozë, mund të ndodhë në tre mënyra: marrje masive e hekurit me gojë, rritje të përthithjes së hekurit gjatë marrjes normale të hekurit dhe prodhim të tepruar ose transfuzion masiv, të shpeshtë të qelizave të kuqe të gjakut.
Në hemokromatozën e trashëguar, hekuri i tepërt zakonisht depozitohet në qelizat parenkimale, ndërsa në hemokromatozën e transfuzionit depozitohet kryesisht në qelizat retikuloendoteliale 1-3.
Hemokromatoza e trashëguar përfshin një grup çrregullimesh gjenetike të karakterizuara nga thithja e rritur e hekurit. Mekanizmi mbizotërues në shumicën e llojeve të hemokromatozës trashëgimore është efekti hepcidin, i cili luan një rol kryesor në homeostazën e hekurit 4-6. Hepsidina sintetizohet kryesisht në hepatocitet dhe kontrollon përqendrimin e hekurit në plazmë duke u lidhur me ferroportin (i quajtur edhe SLC40A1), i vetmi transportues i njohur transmembran i hekurit nga indet e dhuruesve të hekurit. Ferroportina eksporton hekur nga duoden, nga makrofagët dhe hepatocitet.
Në plazmën, hekuri lidhet me trasferrin, kështu që ngopja e hekurit me transferrinë është mesatarisht 35% (vlera mesatare e mëngjesit). Hepsidina pengon çlirimin e hekurit nga makrofagët (nga qelizat e vjetra të kuqe të gjakut dhe ferritin), hepatocitet dhe enterocitet duodenale duke u lidhur me ferroportin. Dhe në mungesë të ferroportinës, prodhimi i hekurit nga enterocitet, hepatocitet dhe makrofagët është i bllokuar. Kështu, hepcidina zvogëlon thithjen e hekurit në zorrë, zvogëlon nivelin e hekurit të lëshuar nga hepatocitet dhe makrofagët, gjë që çon në një nivel të ulët të hekurit në plazmë dhe një rritje të indeve.
Shkaku i hemokromatozës trashëgimore është një mutacion në gjenin HFE. Defekti në gjenin HFE u përshkrua për herë të parë në 1996, që është një mutacion që çon në zëvendësimin e tirozinës me cisteinë në pozicionin e aminoacideve 282 (C282Y). Një mutacion në gjenin HFE shkakton thithjen e rritur të hekurit, përkundër marrjes normale të hekurit. Proteina HFE rregullon prodhimin e hepcidinës. Pacientët me homozigotë të trashëguar hemokromatozë C282Y janë nga 80 në 85% 1, 8.
Ekzistojnë dy mutacione të tjera: njëra është e lidhur me zëvendësimin e aspartatit me histidinë në pozicionin 63 (H63D), dhe e dyta është një zëvendësim i cisteinës me serinë në pozicionin 65 (S65C). Këto mutacione nuk kanë lidhje me sindromën e mbingarkesës së hekurit, përveç nëse C282Y është një pjesë integrale e heterozigozave C282Y / H63D ose C282Y / S65C. Kështu, forma e lidhur me HFE e hemokromatozës trashëgimore mund të verifikohet me një kurs asimptomatik të sëmundjes. Në përputhje me rrethanat, një diagnozë gjenetike mund të zbatohet në pacientët në të cilët hemokromatoza ende nuk është shfaqur në mënyrë fenotipike. Ky grup pacientësh me një predispozitë gjenetike ndaj hemokromatozës. Heterozigotët kanë një rrezik të rritur të zhvillimit të diabetit në krahasim me popullsinë e përgjithshme, mekanizmi i zhvillimit është i panjohur 9-11.
Më parë mendohej se në të gjithë pacientët me defekt të gjenit HFE, me kohë do të zhvillohet një klinikë hemokromatoze. Sidoqoftë, tani është zbuluar se shprehja fenotipike gjendet vetëm në afro 70% të homozigotëve C282Y, dhe më pak se 10% e tyre zhvillojnë mbingarkesë të rëndë hekuri me dëmtim të organeve të brendshme 12, 13.
Tabela tregon klasifikimin e sindromave të mbingarkesës së hekurit në varësi të shkakut të shfaqjes së tij.
Në varësi të shkakut të sëmundjes, pacientët me sindromën e mbingarkesës së hekurit mund të ndahen në 4 grupe: pacientë me hemokromatozë të trashëguar, pacientë me hemokromatozë sekondare të shkaktuar nga shkaqe të ndryshme, dhe një grup i vogël pacientësh, i cili dallohet si "i ndryshëm".
Shkaku i hemokromatozës sekondare është hemokromatoza eritropoetike. Më shpesh kjo ndodh si rezultat i një sëmundjeje themelore të gjakut në të cilën qelizat e kuqe të gjakut kanë një jetëgjatësi më të shkurtër. Ky grup sëmundjesh përfshin aneminë e mungesës së hekurit, talaseminë, aneminë sideroblastike, aneminë kronike hemolitike, aneminë aplastike, aneminë e ndjeshme ndaj piridoksinës, aneminë e piruvate kinazës.
Sindroma e mbingarkesës së hekurit mund të ndodhë në pacientët që marrin transfuzion të zgjatur dhe të shumëfishtë të qelizave të kuqe të gjakut. Siç shihet nga tabela, sëmundje të tjera mjaft të rralla, të tilla si, porfiria, gjithashtu mund të shkaktojnë sindromën e mbingarkesës së hekurit.
Më në fund, marrja e tepërt e hekurit mund të shkaktojë hemokromatozë. Fakti i mirënjohur historik: përdorimi i birrës së bërë në daulle çeliku ishte shkaku i sindromës së mbingarkesës së hekurit. Gjithashtu, një mbidozë e përgatitjeve të hekurit mund të shkaktojë sindromën e mbingarkesës së hekurit. Duhet mbajtur mend që shumë shtesa ushqimore pa recetë përmbajnë hekur në një dozë mjaft të madhe, kështu që përdorimi i tyre i pakontrolluar është i papranueshëm.
Simptomat e sëmundjes varen nga organi që është më i prekur, megjithatë, pothuajse të gjithë pacientët ankohen për dobësi dhe lodhje të konsiderueshme. Nuk ka simptoma specifike të hemokromatozës. Më shpesh, diagnoza bëhet në fazën e sëmundjes, kur tashmë janë prekur disa sisteme. Nga simptomat e para të sëmundjes deri në verifikimin e diagnozës zakonisht zgjat të paktën dhjetë vjet. Në gratë me hemokromatozë, simptomat e sëmundjes manifestohen në një moshë të mëvonshme sesa te burrat, për shkak të humbjes së gjakut në menstruacione, humbjes së "hekurit të nënës" gjatë shtatëzanisë dhe efektit antioksidues të estrogjenit, dhe sëmundja nuk manifestohet klinikisht vetveten para periudhës klimaterike.
Përafërsisht 50% e pacientëve me simptoma të hemokromatozës trashëgimore kanë diabet mellitus, rreziku i shfaqjes së tij rritet ndjeshëm në heterozigotët. Cirroza e mëlçisë është e pranishme në 70% të pacientëve me hemokromatozë. Në këtë grup pacientësh, incidenca e karcinomës hepatocelulare, që është shkaku kryesor i vdekjes, është rritur ndjeshëm.
Dëmtimi i nyjeve me hemokromatozë manifestohet në formën e artralgjisë (zakonisht nyjet e dyta dhe të treta metakarpophangeal). Deformimet e kyçeve me hemokromatozën zakonisht nuk ndodhin, megjithëse ndryshimet degjenerative të nyjeve janë të mundshme. Në këta pacientë, si rregull, kristalet e pirofosfatit të kalciumit mund të gjenden në lëngun sinovial. Characteristicshtë karakteristikë e poliartritit me hemokromatozë që edhe pas normalizimit të depozitave të hekurit, ai akoma mund të përparojë.
Depozitimi i hekurit në fibrat e muskujve të zemrës dhe qelizat e sistemit të përcjelljes së zemrës mund të çojë në shqetësim të ritmit të zemrës dhe / ose kardiomiopati të holluar, me zhvillimin e mëtutjeshëm të dështimit të zemrës. Në disa raste, ekziston një kompensim i plotë për dështimin e ventrikulit të majtë pas normalizimit të nivelit të hekurit në trup 9-12.
Me hemokromatozën, zhvillimi i hipogonadizmit dhe, në përputhje me rrethanat, të pafuqisë për shkak të insuficencës hipotalamike dhe / ose hipofizës, që çon në një shkelje të lëshimit të hormonit gonadotropin, është i mundur. Në rastet e dyqaneve të tepërta të hekurit pesë herë ose më shumë, ndodh hiperpigmentim i lëkurës, që është rezultat i depozitimit të hekurit dhe melaninës. Mbingarkesa e hekurit e makrofagëve mund të çojë në fagocitozë të dëmtuar dhe imunitet të zvogëluar, gjë që çon në një rrezik të shtuar të infeksionit nga Listeria, Yersinia enterocolitica dhe Vibrio vulnificus. Depozitimi i hekurit në gjëndrën tiroide zakonisht shkakton hipotiroidizëm.
Faza e zhvilluar e hemokromatozës karakterizohet nga prania e cirrozës, diabeti mellitus dhe pigmentimi i lëkurës (i ashtuquajturi diabeti i bronzit). Në pacientët që abuzojnë me alkoolin dhe janë të infektuar me hepatit B dhe / ose C, patologjia e mëlçisë dhe pankreasit të shoqëruar me hemokromatozë vazhdon në mënyrë të konsiderueshme më ashpër 1-3.
Diagrami tregon masa diagnostike për hemokromatozë të dyshuar. Dihet se vetëm rreth 70% e homozigotëve C282Y kanë nivele të larta të ferritinës, e cila korrespondon me një rritje në dyqanet e hekurit, dhe vetëm një përqindje e vogël e këtyre pacientëve kanë manifestime klinike të sëmundjes. Sigurisht, të gjithë pacientët me simptoma që mund të shfaqen me hemokromatozë duhet t'i nënshtrohen ekzaminimit të mëtejshëm për të përjashtuar sëmundjen. Vëmendje e veçantë duhet t'i kushtohet pacientëve me dobësi të pamotivuar, artralgji, dhimbje në kuadratin e sipërm të djathtë të barkut, impotencë, rënie të dëshirës seksuale, sindromi i dështimit të zemrës, pigmentim të lëkurës dhe diabeti. Përveç kësaj, në të gjithë pacientët me hepatomegali, sindromë citolitike, me fazën cirotike të sëmundjes, është e nevojshme, përveç të gjitha shkaqeve të mundshme etiologjike të sëmundjes, të mbani mend mundësinë e hemokromatozës. Sigurisht, hemokromatoza trashëgimore duhet të përjashtohet në pacientët me të afërm të shkallës së parë të farefisnisë që vuajnë nga hemokromatoza. 
Studimi duhet të fillojë duke matur ngopjen e koncentrimit të servisit të transfererrinës ose serritit të ferritinës. Duhet të theksohet se përcaktimi i transfererrinës në rastet e hemokromatozës eritropoetike nuk është aq efektive për verifikimin e sindromës së mbingarkesës së hekurit. Specifikimi i ferritinës varet kryesisht nga prania e sëmundjeve inflamatore. Nëse niveli i ferritinës është më i lartë se 200 μg / l në gra ose 300 μg / l tek burrat ose ngopja e transferrinës është më shumë se 40% tek gratë ose 50% tek burrat, testimi i mëtutjeshëm është i nevojshëm për të përjashtuar hemokromatozën 1, 2, 10, 11.
Sipas rekomandimeve të Shoqatës Amerikane për Studimin e Sëmundjeve të Mëlçisë 2011 (AASLD 2011) nëse pacienti ka një transferim serumi prej 1000 mg / l), dhe në varësi të këtyre treguesve, merret një vendim mbi taktikat terapeutike dhe nevojën për biopsi të mëlçisë (shiko grafikun ).
Në pacientët me një kombinim të heterozigotëve C288Y / H63D, si dhe C288Y heterozigot ose jo C288Y, eliminimi i kujdesshëm i sëmundjeve të tjera të mëlçisë ose gjakut është i nevojshëm (nëse është e nevojshme, është e nevojshme një biopsi birë e mëlçisë) dhe më pas merret një vendim për rrjedhjen e gjakut.
Nuk ka asnjë provë të besueshme që dieta të caktuara ndikojnë në fillimin ose përparimin e hemokromatozës. Sidoqoftë, disa autorë besojnë se pacientëve me hemokromatozë trashëgimore u tregohet dietë me përjashtim të çajit dhe agrumeve, të cilat, sipas mendimit të tyre, kontribuojnë në akumulimin e hekurit. Sigurisht, alkooli, i cili është substanca kryesore hepatotoksike, duhet të ndalohet rreptësisht për pacientët me hemokromatozë. Për më tepër, etanoli është provuar që zvogëlon sintezën e hepcidinës 20, 21.
Trajtimi parësor për hemokromatozën parësore është rrjedhja e gjakut. Ulja e numrit të qelizave të kuqe të gjakut, të cilat janë mobilizuesi kryesor i hekurit në trup, duke zvogëluar dhe minimizuar efektin toksik të hekurit. Pacientët mund të kërkojnë 50-100 pllaka gjaku në vit, 500 ml secila, për të ulur nivelin e hekurit në normalitet. Pasi të normalizohet niveli i hekurit, kërkohet rrjedhja e gjakut gjatë gjithë jetës, por më pak e shpeshtë, zakonisht 3-4 herë në vit. Qëllimi i rrjedhjes së gjakut është të ruajë nivelet e ferritinës prej 50-100 µg / L. Në rastet e një rënie të konsiderueshme të hemoglobinës pas rrjedhjes së gjakut, këshillohet trajtimi i përbashkët me eritropoietin.
Nëse hemokromatoza zbulohet në një fazë të hershme të sëmundjes, trajtimi i rrjedhjes së gjakut mund të parandalojë mosfunksionimin e organeve të prekura dhe në këtë mënyrë të rrisë jetëgjatësinë e pacientit. Sidoqoftë, pacientët rrallë jetojnë më shumë se dy vjet pas diagnozës, në rastet e diagnostikimit të vonë në fazën e manifestimeve klinike të hollësishme 22, 23.
Sipas Shoqatës Evropiane për Studimin e Mëlçisë (EASL 2010), indikacionet për gjakderdhjen terapeutike janë nivele të ngritura të ferritinës në serum. Rekomandohet që rrjedhja terapeutike e gjakut me një vëllim prej 400-500 ml të kryhet një herë në javë ose një herë në 2 javë derisa të arrihet një nivel ferritini prej 45% dhe një rritje e konsiderueshme e ferritinës në serum deri në 1444 mcg / l, diagnoza e hemokromatozës është e pamohueshme. Mostrat e ADN-së u analizuan për mutacione në gjenin HFE - një mutacion C282Y (c.845 G> A) u zbulua në gjendjen homozigoze s.845A / s.845 A.
Kështu që, diagnoza e pacientit K. është hemokromatoza trashëgimore, një mutacion homozigoz në gjenin HFE (C288Y / C288Y) me dëmtim mbizotërues të mëlçisë, fibrozë të shkallës 1 (FibroScan, Metavir 6.6 kPa).
Manifestimi i vonë dhe diagnostikimi i sëmundjes në moshën 58 vjeç në 2015 është për shkak të kompensimit afatgjatë të sëmundjes për shkak të humbjes masive të gjakut për shkak të gjakut menstrual, dhurimit të gjakut, humbjes së gjakut gjatë përfundimit të shtatzënisë dhe lindjes së fëmijëve.
Vlen të përmendet që kanë kaluar 8 vjet nga shfaqja e shenjave të para të sëmundjes në verifikimin e diagnozës! Që nga fundi i vitit 2015, pacientit i është përshkruar terapi - gjakderdhje prej 500 ml një herë në javë. Pacienti e toleroi mirë rrjedhjen e gjakut, vuri në dukje një përmirësim të ndjeshëm të gjendjes pas procedurës së parë. U monitorua një test i përgjithshëm i gjakut dhe ferritina e gjakut, niveli i të cilit gradualisht u ul. Në total, më shumë se 100 gjakderdhje janë kryer në 2 vjet, por deri më tani niveli i transferimit të synuar (100 μg / l) nuk është arritur për shkak të pacientit që kalon periodikisht procedurën, duke shpjeguar shëndetin e saj të mirë. Aktualisht, pacienti vazhdon terapinë, ajo arriti ta bindë atë për nevojën e terapisë gjatë gjithë jetës.
Kështu, duhet të mbahet mend se në prani të një sindromi citolitik në pacientë, hemokromatoza trashëgimore duhet të përfshihet në kërkimin diagnostik. Terapia e zgjedhjes për hemokromatozën trashëgimore aktualisht mbetet gjakderdhje. Terapia adekuate e filluar me kohë lejon të shmangni zhvillimin e fazës cirotike të sëmundjes dhe në këtë mënyrë të rrit jetëgjatësinë e pacientëve.
Informacione për autorët:
Voloshina Natalya Borisovna - kandidat i shkencave mjekësore, profesor i asociuar propaedeutika e sëmundjeve të brendshme të fakultetit mjekësor
Osipenko Marina Fedorovna - doktor i shkencave mjekësore, prof. dept. propaedeutika e sëmundjeve të brendshme të fakultetit mjekësor
Voloshin Andrey Nikolaevich - Doktor i Spitalit Klinik të qytetit Novosibirsk 2
Hemokromatoza: cila është kjo sëmundje?
Për të kuptuar thelbin e sëmundjes, duhet të dini se sa hekuri duhet të ketë një person normalisht. Tek burrat, hekuri është rreth 500-1500 mg, dhe tek gratë, nga 300 në 1000 mg. Treguesit varen jo vetëm nga gjinia, por edhe nga pesha e personit. Më shumë se gjysma e sasisë totale të hekurit është në hemoglobinë.
Rreth 20 mg të këtij mikroelementi hyjnë në trup me ushqim në ditë. Nga këto, vetëm 1-1,5 mg thithet në zorrë. Me hemokromatozë (GC) ose siderofili, siç quhet edhe kjo sëmundje, përthithja rritet në 4 mg në ditë, dhe hekuri gradualisht grumbullohet në indet e organeve të ndryshme.
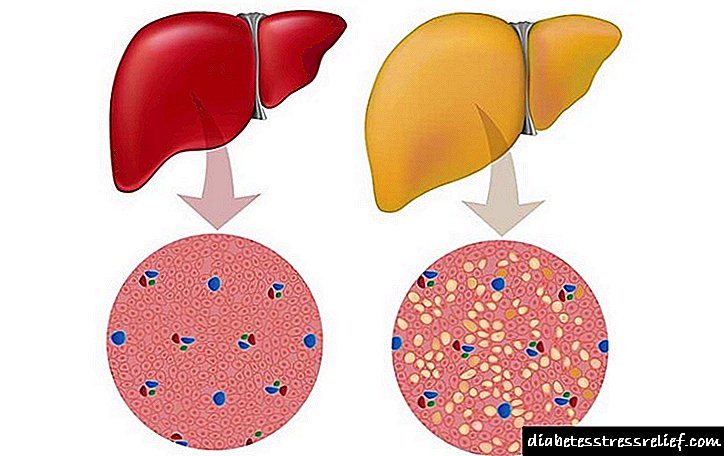
Mëlçia e shëndetshme dhe hemokromatoza
Teprica e saj shkatërron molekulat e proteinave dhe karbohidrateve, dhe kështu vetë organin. Në pacientët me GC, sasia e hekurit në mëlçi mund të arrijë në 1% të masës së thatë të organit, e cila është e mbushur me cirrozë, dhe në një të tretën e rasteve me kancer të mëlçisë. Dëmtuar nga hekuri i tepërt, pankreasi mund të japë impuls në zhvillimin e diabetit.
Duke u depozituar në gjëndrën e hipofizës, hekuri shkatërron të gjithë sistemin endokrin. Organet riprodhuese vuajnë më shumë se të tjerët: burrat kanë mosfunksionim ngrerë dhe gratë mund të zhvillojnë infertilitet.
Shkaqet e shfaqjes
Arsyeja kryesore për GC është “mosfunksionimi” i gjenit, ose më saktë, i gjenit HFE. Shtë ai që rregullon rrjedhën e proceseve kimike dhe sasinë e hekurit që hyn në trup si pjesë e ushqimit. Mutacioni që ndodh në të çon në prishje të metabolizmit të hekurit.
Shkaqe të tjera të GC janë:
- talasemia. Në këtë rast, struktura e hemoglobinës shkatërrohet me lëshimin e hekurit, dmth.
- hepatiti,
- hekuri mund të rritet si rezultat i transfuzioneve të shpeshta të gjakut. Fakti është se jeta e qelizave të kuqe të gjakut të huaj është shumë më e shkurtër se e tyre. Kur vdesin, lëshojnë hekurin,
- procedurat e hemodializës.
Kodi ICD-10 dhe klasifikimi
Në klasifikuesin e pranuar përgjithësisht të sëmundjeve të GC, caktohet kodi E83.1.
Në një vegjë etilogjike, GC parësore (ose GG trashëgimore) dhe dytësore dallohen:
- kryesor. Ky lloj i sëmundjes ka një natyrë të trashëgueshme dhe është rezultat i një defekti në sistemin enzimë që ndikon në metabolizmin e hekurit. Diagnostifikohet në 3 persona nga 1000. Vihet re se burrat janë më të ndjeshëm ndaj kësaj patologjie dhe vuajnë prej saj 3 herë më shpesh sesa gratë,
- dytësor. Shkaku i saj janë sëmundjet e mëlçisë së pacientit (e cila shpesh vërehet me alkoolizëm), transfuzioni i gjakut, vetë-trajtimi me komplekse vitaminash me një përmbajtje të lartë hekuri. Shkaku i GC i fituar mund të jetë problemet e lëkurës dhe sëmundjet e gjakut.
Hemokromatoza primare (PCH) karakterizohet nga një zhvillim gradual, dhe në fazat e hershme, pacientët ankohen për lodhje. Ata mund të shqetësohen nga dhimbja në anën e djathtë dhe lëkura e thatë.
Faza e zgjeruar e PCH karakterizohet nga:
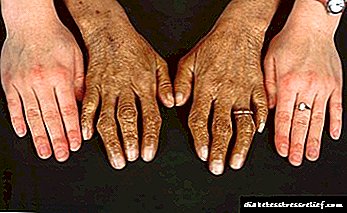
- pigmentim specifik i fytyrës, qafës, krahëve dhe sqetullave. Ata marrin një ngjyrosje bronzi,
- cirroza e mëlçisë. Diagnostifikohet në 95% të rasteve,
- dështimi i zemrës
- artrit,
- diabeti mellitus: në 50% të rasteve,
- shpretkë e zmadhuar,
- mosfunksionimi seksual.
Në fazat e fundit, vërehet hipertensioni portal dhe ascitet. Kanceri i mëlçisë mund të zhvillohet.
 Meqenëse hekuri i tepërt formohet me kalimin e viteve, simptomat fillestare të GC sekondare manifestohen tek burrat pas 40 vjetësh, dhe tek gratë pas 60 vjetësh.
Meqenëse hekuri i tepërt formohet me kalimin e viteve, simptomat fillestare të GC sekondare manifestohen tek burrat pas 40 vjetësh, dhe tek gratë pas 60 vjetësh.
Simptomat janë si më poshtë:
- melasma,
- lodhje dhe humbje peshe,
- ulur epsh
- zmadhimi dhe dendësimi i indeve të mëlçisë,
- cirroza (në fazën e fundit të GC).
Testi i gjakut dhe metoda të tjera diagnostikuese
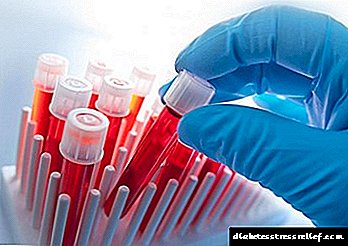 Një gastroenterolog konfirmon diagnozën. Në fazat e hershme të sëmundjes, testet laboratorike janë shumë të rëndësishme.
Një gastroenterolog konfirmon diagnozën. Në fazat e hershme të sëmundjes, testet laboratorike janë shumë të rëndësishme.
Me GC, bëhen teste speciale të gjakut për të zbuluar vlerat e hekurit në plazmë, aftësinë e tij të ulët të lidhjes së hekurit dhe ngopjen me transferrinën.
Shenja kryesore e sëmundjes është depozitimi i hemosiderinës në hepatocitet e mëlçisë, në lëkurë dhe organe të tjera, të cilat bëhen "të ndryshkur" për shkak të tepërt të këtij pigmenti. Një test i përgjithshëm i gjakut kërkohet gjithashtu për biokimi, si dhe sheqer. Përveç kësaj, janë marrë teste të mëlçisë.
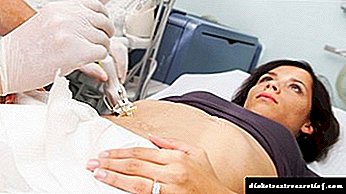 Për më tepër, studime instrumentale janë kryer gjithashtu:
Për më tepër, studime instrumentale janë kryer gjithashtu:
- biopsia e mëlçisë është mënyra kryesore për të konfirmuar GC,
- Ultratinguj i barkut
- MRI e mëlçisë (në disa raste)
- ekokardiografia, për të përjashtuar / konfirmuar kardiomiopatinë,
- radiografi e përbashkët.
Dietë terapeutike
Shtë e rëndësishme të kuptohet që me një hemokromatozë të diagnostikuar, dieta duhet të jetë e gjatë.
Rregulli kryesor është ulja maksimale e dietës së produkteve që përmbajnë hekur, veçanërisht:

- djathra të fortë dhe peshq deti,
- drithëra: tërshëra, meli dhe hikërror,
- bukë e zezë
- bishtajore dhe fruta të thata,
- acid askorbik dhe ilaçe me një përmbajtje të lartë të vitaminës C,
- offal, veçanërisht mëlçia, është plotësisht e përjashtuar.
Alkooli është një tabu absolute. Por, çaji dhe kafja, përkundrazi, tregohen. Ata kanë tanin, i cili ngadalëson thithjen e hekurit.
Lista e barnave të përdorura
Ky trajtim kryhet me ilaçe që largojnë hekurin nga trupi i pacientit. Në fazën fillestare, përshkruhen vitaminat A, E dhe acidi folik. Pastaj përdoren chelators (të tilla si Desferal).

Doza e injektimit: 1g / ditë. Tashmë 500 mg të ilaçit jep një rezultat të prekshëm: ekskretohet deri në 43 mg hekur. Kursi zgjat deri në 1.5 muaj. Përdorimi i zgjatur është i rrezikshëm: kapja e lenteve është e mundur.
Flebotomia dhe procedurat e tjera terapeutike
 Flebotomia është trajtimi më i thjeshtë dhe, në të njëjtën kohë, mjaft efektiv jo-farmakologjik i GC.
Flebotomia është trajtimi më i thjeshtë dhe, në të njëjtën kohë, mjaft efektiv jo-farmakologjik i GC.
Një birë bëhet në venë e pacientit dhe gjaku lëshohet nga trupi. Kullohen rreth 500 ml në javë.
Procedura kryhet vetëm mbi baza ambulatore. Gjaku testohet vazhdimisht për përqendrimin e ferrinës: duhet të bjerë në 50. Kjo mund të zgjasë 2-3 vjet. Më tej, terapia ka për qëllim ruajtjen e vlerës optimale të këtij elementi gjurmë.
Trajtimi me mjete juridike popullore
Kjo terapi ka një efekt të butë në organet e sëmura.
Trajtimi i mëlçisë:

- kungull. Goodshtë mirë si i papërpunuar ashtu edhe i pjekur. Perimet shtohen në sallata ose përzihen me mjaltë - të shijshme dhe të shëndetshme! Tregohet edhe lëngu i kungullit: gjysmë gotë në stomak bosh,
- panxhar- Një produkt tjetër i dobishëm për GC. Përdorni në formë të papërpunuar ose të zier. Lëng i shëndetshëm dhe i shtrydhur fllad.
Për trajtimin e zemrës, mund të këshilloni infuzione të murrizit, adonisit ose nënës. Bimët derdhen me ujë të valë dhe, pasi insistojnë, janë të dehur sipas udhëzimeve.
Trajtimi i pankreasit:

- zierje e farave të gjetheve do të ndihmojë. Përmasat: 1 tbsp. lëndëve të para në 1 tbsp. uji. Farat e pjekura zihen për 5 minuta, ftohen dhe merren para ngrënies, 1 lugë gjelle.,
- mjaltë me kanellë. Përmasat: 1 tbsp. pluhur në 1 lugë gjelle ujë. Këmbëngulni 15-30 minuta. dhe shtoni pak mjaltë. Lëreni edhe për 2 orë të tjera. Të gjitha mjetet duhet të pihen brenda një dite.
Bollgur e dobishme dhe e pjekur (me lëvore). Përmasat: 100 g drithëra në 1,5 litër ujë. Vlim për të paktën gjysmë ore. Më pas, menjëherë në tasin ku janë gatuar tërshërët, shtypeni derisa të zihet furra dhe ziej përsëri për 40 minuta. Jeta e supës së filtruar nuk është më shumë se 2 ditë. Pini gjysmë gotë para ngrënies.
Prognoza dhe udhëzimet kryesore klinike
Por nëse terapia kryhet nën mbikëqyrjen mjekësore dhe me kohë, atëherë jeta e pacientit rritet ndjeshëm.
Duke qenë një sëmundje trashëgimore, hemokromatoza në 25% të rasteve diagnostikohet në të afërmit e pacientit. Pra, ato duhet të ekzaminohen më tej. Kjo do të zbulojë sëmundjen edhe para manifestimeve klinike dhe në të ardhmen për të shmangur komplikimet e saj.
Në rastin e GC sekondar, rekomandohet dieta, është e rëndësishme të mbani nën kontroll gjendjen e mëlçisë dhe gjakut. Hemokromatoza e zbuluar gjatë shtatëzënësisë (ose në fazën e planifikimit) nuk është e rrezikshme.
Video të lidhura
Në lidhje me simptomat, shkaqet dhe metodat e trajtimit të hemokromatozës në video:
Fatkeqësisht, shkaku kryesor i hemokromatozës nuk është identifikuar ende. Por aktualisht, një teknikë e veçantë e plotë e trajtimit është zhvilluar dhe është përdorur në mënyrë aktive, qëllimi i së cilës është të ndërpresë manifestimet klinike të sëmundjes dhe të zvogëlojë rrezikun e komplikimeve të mundshme të tij.
- Stabilizon nivelet e sheqerit për një kohë të gjatë
- Rivendos prodhimin e insulinës pankreatike
Mësoni më shumë Jo ilaç. ->
Terapia bashkëkohore e sëmundjes
Hekuri i tepërt në organe çon në zhvillimin e patologjive të shumta. Të gjithë kërkojnë terapi ndihmëse. Për shembull, nëse GC ka kontribuar në zhvillimin e diabetit, kjo e fundit duhet të mjekohet, duke mbajtur gjithmonë nën kontroll nivelin e sheqerit.
Nëse zbulohen patologji në mëlçi, trajtimi i tij është duke vazhduar. Kjo është e nevojshme për të parandaluar zhvillimin e patologjisë në gjendjen e një tumori malinj.
Hemochromatosis
Hemokromatoza e trashëguar (NG) është një sëmundje polististike e bazuar në çrregullime metabolike të përcaktuara gjenetikisht të hekurit, që çon në akumulimin e saj të tepërt në trup dhe dëmtimin toksik të organeve dhe indeve.
Përshkrimi i parë i sëmundjes i përket A. Trousseau (1865), i cili identifikoi një treshe të manifestimeve kryesore klinike: diabet mellitus, pigmentim të lëkurës bronzi, cirrozë. Termi "hemokromatozë" u propozua në 1889 nga F.D. von Recklinghausen. Që nga viti 1935, sëmundja i përket grupit të sëmundjeve trashëgimore. Në vitin 1996, J.N. Feder et al. identifikuar gjenin për hemokromatozën trashëgimore (HFE), mutacionet e të cilit më së shpeshti çojnë në zhvillimin e kësaj sëmundje. Në vitet 2000-2004 përshkruhen mutacionet e gjeneve të tjera që çojnë në zhvillimin e hemokromatozës.
Prevalenca e sëmundjes ndryshon nga 1: 250 individë që jetojnë në Evropën Veriore në 1: 3300 në mesin e popullatës së zezë të Sh.B.A dhe vendeve afrikane. Sëmundja diagnostikohet në meshkuj 5-10 herë më shpesh sesa tek gratë. Gjatë ekzaminimit gjenetik, u zbulua se një mutacion homozigues i gjenit HFE është zbuluar në 1 nga 500 pacientë të ekzaminuar, ndërsa numri i rasteve të përcaktuara klinikisht të NG është 1: 5,000. Kështu, një numër i konsiderueshëm i rasteve të sëmundjes nuk njihen ose diagnostikohen vonë, në fazën e dëmtimit të pakthyeshëm të brendshëm. organet (cirroza, diabeti mellitus, kardiomiopatia e dilatuar).
Në përputhje me bazën gjenetike të sëmundjes, dallohen 4 lloje të hemokromatozës trashëgimore:
Tipi I - i trashëguar nga një mekanizëm autosomal recesiv, për shkak të mutacioneve në gjenin HFE të vendosur në kromozomin 6. Më shpesh (në 87-90% të pacientëve), mutacioni C282Y është regjistruar - zëvendësimi i cisteinës me tirozinë në aminoacidin 282-të. Mutacioni H63D është më pak i zakonshëm - zëvendësimi i citidinës me guaninë në aminoacidin e 63-të,
Lloji II - hemokromatoza e të miturve është e rrallë, për shkak të mutacioneve në gjenin përgjegjës për sintezën e një proteine tjetër të metabolizmit të hekurit - hepsidin, etj.
Lloji III - baza gjenetike përbëhet nga mutacione të një gjenetimi që kodon sintezën e receptorit të transferrinës
Tipi IV - baza gjenetike përbëhet nga mutacione në gjenin SLC40A1, i cili kodon sintezën e proteinës së transportit ferroportin.
Etiologjia dhe patogjeneza
Hekuri është një përbërës i domosdoshëm biokimik i proceseve metabolike më të rëndësishme, nga njëra anë, dhe është një element potencialisht toksik që mund të shkaktojë dëme oksiduese të membranave biologjike, proteinave dhe acideve nukleike, nga ana tjetër. Në përputhje me këtë, homeostaza e hekurit në trupin e njeriut është rregulluar fort. Shumica e këtij elementi i nënshtrohet një procesi riciklimi: makrofagët e shpretkës dhe kapjen e mëlçisë dhe shkatërrojnë qelizat e kuqe të gjakut, të degradojnë hemoglobinën dhe të lëshojnë hekurin, i cili lidhet me transfererrin ose ferritin dhe riciklohet. Humbja fiziologjike ditore e hekurit nuk i kalon 1-2 mg dhe kompensohet nga thithja e një sasie ekuivalente të hekurit në traktin gastrointestinal. Nuk ka mekanizma që kontrollojnë eleminimin e hekurit te njerëzit.
Mutacionet e gjeneve përgjegjëse për sintezën e proteinave të përfshira në metabolizmin e hekurit çojnë në një çekuilibër midis marrjes dhe humbjes së hekurit, akumulimit patologjik të këtij elementi në organe dhe inde dhe shfaqjen e hekurit të lirë (jo të shoqëruar me transfererrin) në gjak. Zhvillimi i hemokromatozës së tipit I shoqërohet me një mutacion të gjenit përgjegjës për sintezën e proteinës HFE (proteina hemokromatoze), e cila është një glikoproteinë (MM = 37,235 dalton), e ngjashme në strukturë me proteinat e kompleksit kryesor histokompatibiliteti të klasës 1. Funksioni i proteinës HFE në metabolizmin e hekurit dhe mekanizmi i një rritje të mprehtë të thithjes së hekurit gjatë mutacioneve në gjenin HFE nuk janë vendosur plotësisht.
Patogjeneza e hemokromatozës së tipit II-IV shoqërohet me mutacione në gjenet që kodojnë proteina të tjera të përfshira në metabolizmin e hekurit - hepsidin, receptorin transferrin-II, ferroportin.
Një tipar dallues i tipit IV NG, i cili bazohet në mutacionet e gjenit të ferroportinës, është një shkelje mbizotëruese e proceseve të riciklimit të hekurit, i cili fenotipikisht manifestohet si anemi e thellë hipokromike dhe eritropoizë e mungesës së hekurit në kombinim me hemokromatozën e rëndë të organeve të brendshme.
Akumulimi patologjik i hekurit në organet parenkimale shoqërohet me ndryshime degjenerative në parenkimën e qelizës dhe zhvillimin progresiv të indeve fibroze, gjë që çon në mosfunksionim të pakthyeshëm të organeve vitale. Organet më të rrezikuara të synuara janë mëlçia, zemra dhe pankreasi.
Shenjat dhe simptomat klinike
Fotografia klinike e NG përcaktohet nga niveli i akumulimit të hekurit në organe dhe inde. Me hipertension tip I, manifestimet klinike zakonisht gjenden në moshën 45-50 vjeç dhe më të vjetër. Në hemokromatozën e të miturve (lloji II), lezione të rënda të mëlçisë dhe zemrës shfaqen herët - në dekadën e dytë ose të tretë të jetës. Tek burrat, manifestimet klinike të sëmundjes vërehen 3 herë më shpesh sesa tek gratë, gjë që shoqërohet me karakteristikat fiziologjike të trupit të femrës. Manifestimet kryesore klinike përfshijnë simptoma të dëmtimit të mëlçisë, zemrës, organeve të sistemit endokrin dhe nyjeve.
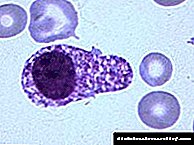 Shenjat e dëmtimit të mëlçisë mund të zbulohen gjatë një ekzaminimi të rastit në formën e një rritjeje pa lëvizje të transaminazave ose debutimit me simptoma të hipertensionit portal: ascitet, hepatosplenomegaly, gjakderdhje nga venat me variçe të ezofagut dhe stomakut.
Shenjat e dëmtimit të mëlçisë mund të zbulohen gjatë një ekzaminimi të rastit në formën e një rritjeje pa lëvizje të transaminazave ose debutimit me simptoma të hipertensionit portal: ascitet, hepatosplenomegaly, gjakderdhje nga venat me variçe të ezofagut dhe stomakut.
Simptomat e dëmtimit të zemrës përfshijnë sulmet në zemër, zhvillimin e aritmive dhe shenjat e dështimit të zemrës. Kardiomiopatia e rëndë është shkaku kryesor i vdekjes tek pacientët e rinj.
Zhvillimi i diabetit dhe mosfunksionimi i gjëndrave gjenitale janë simptoma karakteristike e NG. Tek burrat shpesh vërehet atrofia e testikujve, ulja e shtytjes së seksit, impotenca, azoospermia, tek gratë - amenorrea, infertiliteti.
Dëmtimi i nyjeve manifestohet nga artralgjia e vazhdueshme, nyjet metakarpophalangeal janë më shpesh të përfshira, më rrallë nyjet e gjurit, hip dhe bërryl. Ngurtësia e nyjeve zhvillohet gradualisht.
Manifestime të tjera klinike të NG përfshijnë dobësi të dukshme të pa lëvizshme, lodhje, përgjumje, periudha të dhimbjes së barkut me intensitet të ndryshëm dhe lokalizim, hiperpigmentim të lëkurës dhe një tendencë ndaj infeksioneve të ndryshme (përfshirë mikroorganizmat që rrallë prekin njerëzit e shëndetshëm - Yersenia enterocolitica dhe Vibrio vulnificus).
Diagnoza e NG përcaktohet në bazë të një tabloje karakteristike klinike dhe laboratorike.Shtë e lehtë të dyshosh për diagnozën e hemokromatozës në një pacient me një kombinim të simptomave të mëposhtme: artralgji, dhimbje barku, lëkurë bronzi-gri, prani të diabeti mellitus dhe hepatomegaly.
Testi i gjakut: një kombinim i një niveli të lartë të hemoglobinës me një përqendrim të ulët të hemoglobinës në eritrocitet (MCH) është karakteristik. Zhvillimi i anemisë ose citopenisë tjetër vërehet në fazat e vona të sëmundjes - te pacientët me cirrozë të mëlçisë, ose është rezultat i rrjedhjeve të shumta të gjakut.
Studimi i metabolizmit të hekurit e domosdoshme për të identifikuar shenjat laboratorike të mbingarkesës së hekurit dhe përfshin përcaktimin e nivelit të hekurit, ferritinës dhe transferimit të serumit të gjakut, kapacitetit total të lidhjes së hekurit në serum (OZHSS) dhe koeficientit të vlerësuar të ngopjes së transferimit të hekurit (NTZH). NG karakterizohet nga një rritje në nivelet e hekurit dhe ferritinës në serum, një rënie e niveleve të OGSS dhe transferrinës. Një shenjë e rëndësishme laboratorike e hemokromatozës është një rritje në koeficientin e IST tek burrat mbi 60%, tek gratë - mbi 50%.
Test desferal konfirmon praninë e mbingarkesës së hekurit: pas 0.5 g intramuskulare deferoxamine (desferal), sekretimi ditor i hekurit në urinë tejkalon ndjeshëm nivelin normal (0-5 mmol / ditë).
Në llojin IV NG, pamja laboratorike mund të përfaqësohet nga anemia e thellë hipokromike, hipospiderinemia dhe ferritina e serumit të ngritur, e cila është e kombinuar me mbingarkesë të rëndë të indeve me hekur.
Kryerja e analizës gjenetike molekulare ju lejon të konfirmoni natyrën trashëgimore të hemokromatozës dhe të përjashtoni natyrën sekondare të mbingarkesës së hekurit. Diagnoza e NG është vendosur në prani të mutacioneve homozigoze të gjenit HFE (C282Y ose H63D) ose kur heterozigotet komplekse (një kombinim i mutacioneve heterozigoze C282Y dhe H63D) zbulohen në pacientët me shenja laboratorike të mbingarkesës së hekurit. Mutacionet heterozigore të izoluara C282Y dhe H63D gjenden në popullatën e personave të shëndetshëm me një frekuencë prej 10.6% dhe 23.4% të rasteve, përkatësisht, prania e këtyre mutacioneve nuk është baza për diagnozën e NG.
Skanimi CT i organeve të barkut zbulon një densitet të rritur të indeve të mëlçisë për shkak të depozitave të hekurit dhe lejon që të dyshohet prania e hemokromatozës.
Me MRI mëlçia e një pacienti me hemokromatozë ka një ngjyrë gri të errët ose të zezë. CT dhe MRI e mëlçisë janë të domosdoshme për të përjashtuar diagnozën e karcinomës hepatocelulare.
Biopsia e mëlçisë me një përcaktim gjysmë sasior ose sasior të përmbajtjes së hekurit ju lejon të përcaktoni shkallën e zhvillimit të fibrozës dhe përqendrimin e hekurit në indet e mëlçisë. Për diagnozën e hemokromatozës, rekomandohet të llogaritet “indeksi hepatik hepatik”, i cili është i barabartë me raportin e përmbajtjes së hekurit në indet e mëlçisë (në peshë të thatë mikromol / g) me moshën e pacientit (në vite). Një indeks> 2.0 konfirmon diagnozën e NG.
Hemokromatoza e trashëguar duhet të diferencohet me sindromat sekondare të mbingarkesës së hekurit, të cilat zhvillohen tek pacientët me anemi hemolitike të trashëguar dhe të fituar, disa forma të sindromës myelodysplastic (anemi sideroblastike refraktare), porfiria, si dhe te pacientët me dëmtim të mëlçisë alkoolike.
Qëllimi i trajtimit të NG është largimi i hekurit të tepërt nga trupi dhe parandalimi i dëmtimit të pakthyeshëm të organeve të brendshme. Një metodë e zakonshme e trajtimit është rrjedhja e gjakut. Kursi fillestar konsiston në gjakderdhje në një vëllim prej 500 ml një herë në javë. Pas uljes së nivelit të hemoglobinës me 15-20 g / l, niveli i MCV me 3-5 fl. dhe përmbajtja e ferritinës në serum deri në 20-50 ng / ml, shkoni në terapinë e mirëmbajtjes - heqja e 500 ml gjak çdo 2-4 muaj tek burrat dhe çdo 3-6 muaj në gratë. Trajtimi është i përjetshëm.
Në prani të anemisë ose kundërindikacioneve të tjera (për shembull, dështimi i zemrës), grumbulluesit e hekurit përdoren për rrjedhjen e gjakut. Deferoxamina lidh hekurin e tepërt në inde dhe serum të gjakut dhe ekskreton me urinë dhe feces. Sidoqoftë, gjysma e jetës së këtij ilaçi është e shkurtër - vetëm 10 minuta, e cila kërkon administrim të ngadaltë: intravenozisht në formën e infuzioneve 3-4 orë ose nënlëkurës, mundësisht në formën e infuzioneve 12-orëshe ose gjatë gjithë kohës duke përdorur pompa speciale. Barnat e reja që formojnë komplekse për administrim oral janë zhvilluar dhe janë në fazën e studimit ose zbatimit klinik, nga të cilët më efektivi është Deferasirox.
Efektiviteti i trajtimit përcaktohet nga dinamika e të dhënave klinike dhe laboratorike. Gjendja e pacientëve fillon të përmirësohet pas një kursi të rrjedhjes së gjakut: dobësia, lodhja, përgjumja zhduken, madhësia e mëlçisë zvogëlohet, kursi i diabetit dhe kardiomiopatia mund të përmirësohen. Kontrolli laboratorik përfshin studimin e hemogramit, treguesit e ferritinës, hekurit dhe NTZH (1 herë në 3 muaj), nivelin e sekretimit të hekurit urinar.
Në rast të diagnostikimit të hershëm të hipertensionit dhe rrjedhjes së gjakut terapeutike në kohë, prognoza është e favorshme: jetëgjatësia e pacientëve nuk ndryshon nga jetëgjatësia e njerëzve që nuk vuajnë nga hemokromatoza. Në rastet e diagnostikimit të vonë të sëmundjes, në prani të cirrozës së mëlçisë, kardiomiopatisë, diabet mellitus, prognoza përcaktohet nga ashpërsia e këtyre komplikimeve të pakthyeshme. Shkaqet kryesore të vdekjes së pacientëve janë: komplikimet e diabetit, dështimi i zemrës, kanceri primar i mëlçisë, dështimi i mëlçisë, gjakderdhja nga venat me variçe të ezofagut dhe stomakut, infeksione ndërkurrente.
Informacion i përgjithshëm
Hemokromatoza (diabeti i bronzit, cirroza pigmentare) është një shkelje e shkaktuar gjenetikisht e metabolizmit të hekurit, duke çuar në depozitimin e pigmenteve që përmbajnë hekur në inde dhe organe dhe zhvillimin e dështimit të shumëfishtë të organeve. Sëmundja, e shoqëruar nga një kompleks simptomë simptomash (pigmentim i lëkurës, cirrozë e mëlçisë dhe diabeti mellitus) u përshkrua në 1871, dhe në 1889 u quajt hemokromatoza për ngjyrën karakteristike të lëkurës dhe organeve të brendshme. Frekuenca e hemokromatozës trashëgimore në një popullatë është 1.5-3 raste për 1000 popullatë. Burrat vuajnë nga hemokromatoza 2-3 herë më shpesh se gratë. Mosha mesatare e zhvillimit të patologjisë është 40-60 vjet. Për shkak të natyrës polisistike të lezionit, disiplina të ndryshme klinike janë të përfshira në studimin e hemokromatozës: gastroenterologji, kardiologji, endokrinologji, reumatologji, etj.
Në aspektin etiologjik, dallohen hemokromatoza primare (trashëgimore) dhe sekondare. Hemokromatoza primare shoqërohet me një defekt në sistemet enzimë, gjë që çon në depozitimin e hekurit në organet e brendshme. Në varësi të defektit të gjenit dhe figurës klinike, dallohen 4 forma të hemokromatozës trashëgimore:
- I - autosomik klasik recesiv, tip i lidhur me HFE (më shumë se 95% të rasteve)
- II - lloji i të miturve
- III - lloji i trashëguar HFE-trashëgues (mutacionet në receptorin e transferimit të tipit 2)
- IV– lloji autosomal mbizotërues.
Hemokromatoza sekondare (hemosideroza e përgjithësuar) zhvillohet si rezultat i pamjaftueshmërisë së fituar të sistemeve enzimë të përfshira në metabolizmin e hekurit, dhe shpesh shoqërohet me sëmundje të tjera, në lidhje me të cilat dallohen variantet e saj të mëposhtme: post-transfuzion, ushqyes, metabolik, i përzier dhe neonatal.
Në rrjedhën klinike, hemokromatoza kalon nëpër 3 faza: I - pa mbingarkesë hekuri, II - me mbingarkesë hekuri, por pa simptoma klinike, III - me zhvillimin e manifestimeve klinike.

Shkaqet e hemokromatozës
Hemokromatoza primare e trashëguar është një çrregullim autosomal recesiv i transmetimit. Bazohet në mutacionet e gjenit HFE të vendosura në krahun e shkurtër të kromozomit të 6-të. Një defekt në gjenin HFE çon në prishjen e marrjes së hekurit të ndërmjetësuar nga transfererrina nga qelizat e duodenit 12, duke rezultuar në formimin e një sinjali të rremë për mungesën e hekurit në trup. Nga ana tjetër, kjo kontribuon në rritjen e sintezës së proteinës lidhëse të hekurit DCT-1 nga enterocitet dhe thithjen e rritur të hekurit në zorrë (me marrjen normale të elementëve gjurmë nga ushqimi). Në të ardhmen, ekziston një depozitim i tepërt i pigmentit të hemosiderinës që përmban hekurin në shumë organe të brendshme, vdekja e elementeve të tyre funksionalisht aktive me zhvillimin e proceseve sklerotike. Me hemokromatozën, 0.5-1.0 g hekur grumbullohen çdo vit në trupin e njeriut, dhe manifestimet e sëmundjes manifestohen kur arrihet niveli i përgjithshëm i hekurit prej 20 g (nganjëherë 40-50 g ose më shumë).
Hemokromatoza sekondare zhvillohet si rezultat i marrjes së tepruar ekzogjene të hekurit në trup. Kjo gjendje mund të ndodhë me transfuzione të shpeshta të gjakut të përsëritura, marrje të pakontrolluar të përgatitjeve të hekurit, talasemisë, disa lloje të anemisë, porfirisë së lëkurës, cirrozës alkoolike të mëlçisë, hepatiti kronik viral B dhe C, neoplazma malinje, pas një diete me proteina të ulët.
Simptomat e hemokromatozës
Manifestimi klinik i hemokromatozës trashëgimore ndodh në moshën madhore, kur përmbajtja totale e hekurit në trup arrin vlera kritike (20-40 g). Në varësi të sindromave mbizotëruese, dallohen hepatopatike (hemokromatoza e mëlçisë), kardiopatike (hemokromatoza e zemrës), format endokrinologjike të sëmundjes.
Sëmundja zhvillohet gradualisht, në fazën fillestare ankesat jo-specifike mbizotërojnë për lodhje të shtuar, dobësi, humbje peshe, ulje të dëshirës seksuale. Në këtë fazë, pacientët mund të shqetësohen nga dhimbja në hipokondriumin e duhur, lëkura e thatë, arthralgia për shkak të kondrocalcinosis të nyjeve të mëdha. Në fazën e zgjeruar të hemokromatozës, formohet një kompleks klasik i simptomave, i përfaqësuar nga pigmentimi i lëkurës (lëkura bronzi), cirroza, diabeti mellitus, kardiomiopatia, hipogonadizmi.
Zakonisht, shenja më e hershme e hemokromatozës është shfaqja e një ngjyre specifike të lëkurës dhe mukozave, e shprehur kryesisht në fytyrë, qafë, gjymtyrët e sipërme, në sqetullat dhe organet gjenitale të jashtme, dhe plagët e lëkurës. Intensiteti i pigmentimit varet nga kohëzgjatja e rrjedhës së sëmundjes dhe ndryshon nga gri i zbehtë (i tymosur) deri në bronz-kafe. Karakteristikë është humbja e flokëve në kokë dhe në bagazhin, deformim konkav (në formë lugë) të thonjve. Arthropatitë e nyjeve metakarpofalangeal, nganjëherë në gju, hip dhe bërryl shënohen me zhvillimin e mëvonshëm të ngurtësisë së tyre.
Pothuajse në të gjithë pacientët zbulohet një rritje e mëlçisë, splenomegalisë, cirrozës së mëlçisë. Mosfunksionimi i pankreasit shprehet në zhvillimin e diabetit mellitus të varur nga insulina. Si rezultat i dëmtimit të gjëndrës së hipofizës gjatë hemokromatozës, funksioni seksual vuan: te burrat, zhvillohet atrofia testikulare, impotenca, gjinekomastia, tek gratë - amenorre dhe infertilitet. Hemokromatoza e zemrës karakterizohet nga kardiomiopatia dhe ndërlikimet e saj - aritmia, dështimi kronik i zemrës, infarkti i miokardit.
Në fazën terminale të hemokromatozës, zhvillohet hipertensioni portal, ascitet, cachexia. Vdekja e pacientëve, si rregull, ndodh si rezultat i gjakderdhjes nga venat me variçe të ezofagut, dështimi i mëlçisë, dështimi akut i zemrës, koma diabetike, peritoniiti aseptik, sepsë. Hemokromatoza rrit ndjeshëm rrezikun e shfaqjes së kancerit të mëlçisë (karcinoma hepatocelulare).
Diagnoza e hemokromatozës
Në varësi të simptomave mbizotëruese, pacientët me hemokromatozë mund të kërkojnë ndihmë nga specialistë të ndryshëm: një gastroenterolog, kardiolog, endokrinolog, gjinekolog, urolog, reumatolog dhe dermatolog. Ndërkohë, diagnoza e sëmundjes është e njëjtë për variantet e ndryshme klinike të hemokromatozës. Pas vlerësimit të shenjave klinike, pacientëve u caktohet një seri studimesh laboratorike dhe instrumentale për të verifikuar vlefshmërinë e diagnozës.
Kriteret laboratorike për hemokromatozën janë një rritje e konsiderueshme e nivelit të hekurit, ferritinës dhe transferrinës në serumin e gjakut, një rritje në sekretimin e hekurit në urinë dhe një rënie në aftësinë totale të lidhjes së hekurit në serumin e gjakut. Diagnoza konfirmohet me biopsi shpimi të mëlçisë ose lëkurës, në mostrat e të cilave është zbuluar depozitimi i hemosiderinës. Natyra trashëgimore e hemokromatozës përcaktohet si rezultat i diagnostikimit gjenetik molekular.
Për të vlerësuar ashpërsinë e dëmtimit të organeve të brendshme dhe prognozën e sëmundjes, po studiohen testet e mëlçisë, nivelet e glukozës në gjak dhe urinë, hemoglobina glikoziluar, etj. Diagnoza laboratorike e hemokromatozës plotësohet me studime instrumentale: radiografi të përbashkët, EKG, ekokardiografi, ultratinguj të zgavrës së barkut, MRI, etj.
Trajtimi i hemokromatozës
Qëllimi kryesor i terapisë është heqja e hekurit të tepërt nga trupi dhe parandalimi i zhvillimit të komplikimeve. Pacientët me hemokromatozë u përshkruhet një dietë që kufizon ushqimet e larta në hekur (mollë, mish, mëlçi, hikërror, spinaq, etj.), Karbohidratet lehtësisht të tretshëm. Ndalohet marrja e multivitaminave, acidi askorbik, shtesave dietike që përmbajnë hekur, alkool. Për të hequr hekurin e tepërt nga trupi, ata drejtohen në rrjedhjen e gjakut nën kontrollin e hemoglobinës, hematokritit dhe ferritinës. Për këtë qëllim, mund të përdoren metoda hemokorrecionale ekstrakorporale - plazmafereza, hemosorbimi, citapereza.
Terapia patogjenetike me ilaçin e hemokromatozës bazohet në administrimin intramuskular ose intravenoz të joneve Fe3 + që lidhin deferoksaminën tek një pacient. Në të njëjtën kohë, bëhet trajtimi simptomatik i cirrozës së mëlçisë, dështimi i zemrës, diabeti mellitus dhe hipogonadizmi. Me artropati të rëndë përcaktohen indikacionet për artroplastinë (endoprostetikë të nyjeve të prekura). Në pacientët me cirrozë, çështja e transplantimit të mëlçisë po adresohet.
Parashikimi dhe parandalimi i hemokromatozës
Përkundër rrjedhës progresive të sëmundjes, terapia në kohë mund të zgjasë jetën e pacientëve me hemokromatozë për disa dekada. Në mungesë të trajtimit, jetëgjatësia mesatare e pacientëve pas diagnostikimit të patologjisë nuk kalon 4-5 vjet. Prania e komplikimeve të hemokromatozës (kryesisht cirroza e mëlçisë dhe dështimi konjuktiv i zemrës) është një shenjë prognostikisht e pafavorshme.
Me hemokromatozën e trashëguar, parandalimi zbret në kontrollimin familjar, zbulimin e hershëm dhe trajtimin e sëmundjes. Ushqimi racional, monitorimi i administrimit dhe administrimit të përgatitjeve të hekurit, transfuzionit të gjakut, refuzimi i pirjes së alkoolit dhe monitorimi i pacientëve me sëmundje të mëlçisë dhe sistemit të gjakut bëjnë të mundur shmangien e zhvillimit të hemokromatozës sekondare.




-
Shkaqet e diabetit
Shkaqet e diabetit janë të dukshme dhe të fshehura.Së pari duhet të vendosni - a duhet të dini shkakun e shfaqjes (manifestimit) të sëmundjes tuaj? Ndoshta ju personalisht nuk keni nevojë për të, por mjeku që merr pjesë është jetik. ... -

-

-